




Sterilization is intended to render Reusable Medical Devices free from viable mircroorganisms
![]() Prion are not microorganisms and are more resistant than conventional microorganisms.
Prion are not microorganisms and are more resistant than conventional microorganisms.
Sterilization of RMD is based on 3 key concepts
- Sterility Assurance Level (SAL) : It is not possible to systematically control the total absence of viable microrganism. The objective of sterilization is to limit the probability of microorganism survival to a very low level. For RMD’s the probability or Sterility Assurance Level (SAL) is expressed as: not more than 1 viable microorganisms in an amount of one million sterilized items (or 10-6 SAL ).
- Overkill : The quantity, nature and location of microorganisms which may be present on an RMD after cleaning is not known. RMD sterilization processes must demonstrate their ability to inactivate a highly concentrated inoculum of > 1 million of test microorganisms. Test microorganisms are selected for their high resistance to the sterilization process. This conservative margin above the highest level of real life contamination is called overkill method.
- Compatibility: RMD’s remains fully functional and safe for use after sterilization. Compatibility is checked by RMD Manufacturer, which determines the maximal number of sterilization cycles to which an RMD can be exposed before being discarded or repaired.
Various methods are proposed by international standards to demonstrate the capability of a given sterilization process to reach the SAL using overkill method. One of them is the half cycle overkill method, described as follows:
Tests are performed with microorganisms known for their high resistance to the sterilization process (usually bacterial spores).
Once the process has been characterized, it must be verified that an RMD can be efficiently sterilized. The 106 inoculum is placed at the position determined as the most difficult to sterilize on or within an RMD. RMD's are packaged and inserted in a representative challenging load. | 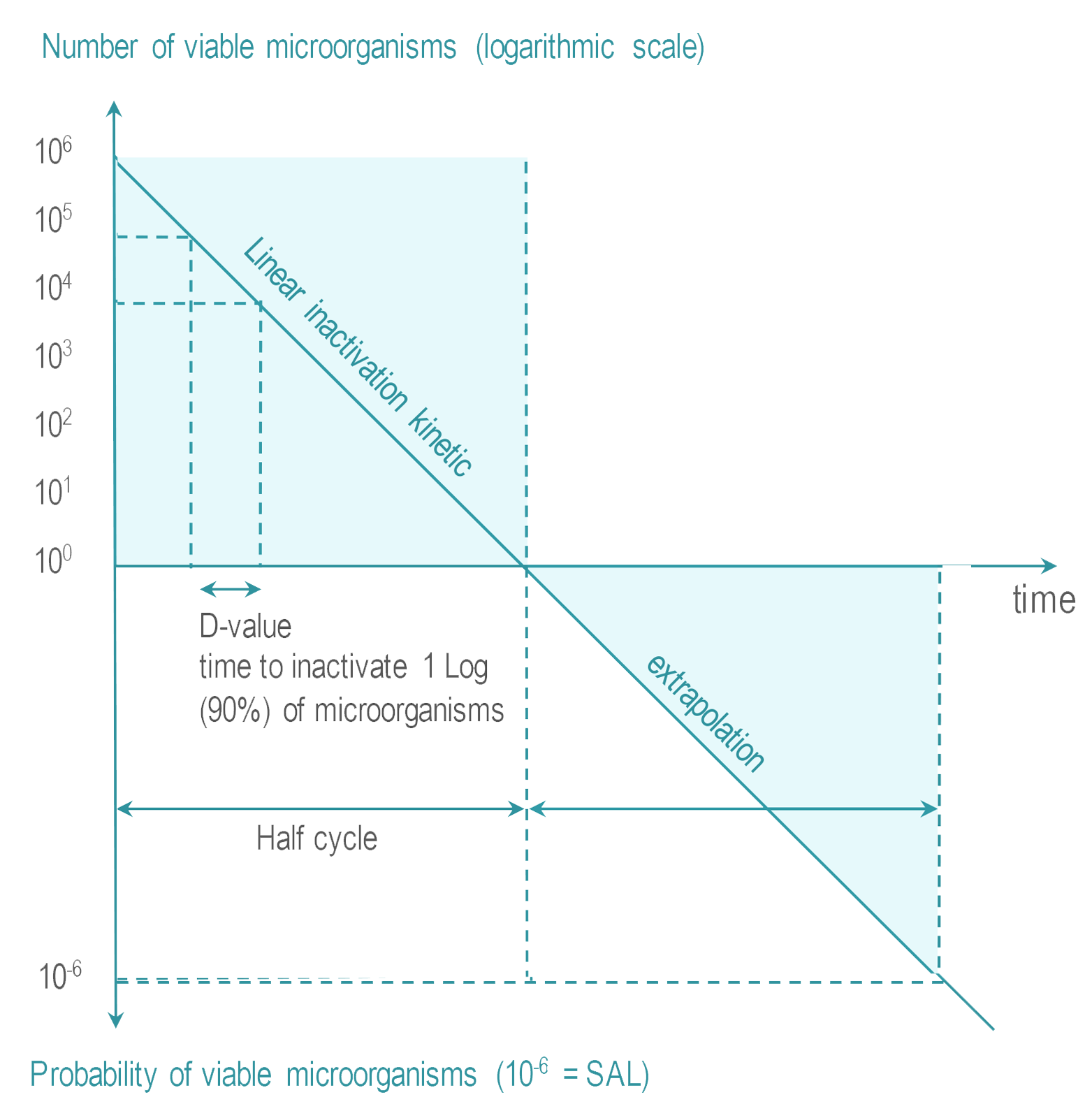 |
Steam sterilization is the most common sterilization method. It is also referred to as moist heat sterilization or saturated steam sterilization),
In dry heat sterilization, RMD are exposed to dry, hot air. Low temperature sterilization (LTS) is adapted tor RMD's, which do not withstand high temperatures. Current low temperature sterilizing agents are: Ethylene Oxide (EtO), steam formaldehyde (LTSF), vaporized hydrogen peroxide (VH2O2). RMD are exposed in controlled temperature, humidity and/or pressure conditions to a minimum concentration (Cc) of sterilizing agent during the time required for achieving the desired SAL.
|
![]() Radiation sterilization (ionizing – gamma, e-beam or high energy X-Ray, or non-ionizing ultra-violet (UV) ) is not commonly used for reprocessing of RMD’s in healthcare facilities and will not be discussed in present guidelines.
Radiation sterilization (ionizing – gamma, e-beam or high energy X-Ray, or non-ionizing ultra-violet (UV) ) is not commonly used for reprocessing of RMD’s in healthcare facilities and will not be discussed in present guidelines.
Non-terminal sterilization methods meet the SAL criteria. However, unlike terminal sterilization, RMD’s are not protected by a packaging. Immediate use steam sterilization (previously called flash sterilization) is an example of non-terminal sterilization process .
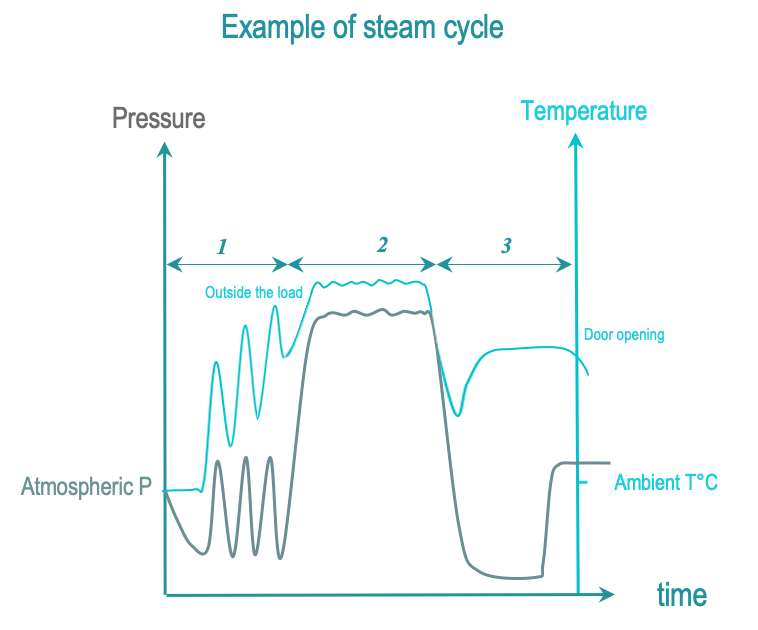
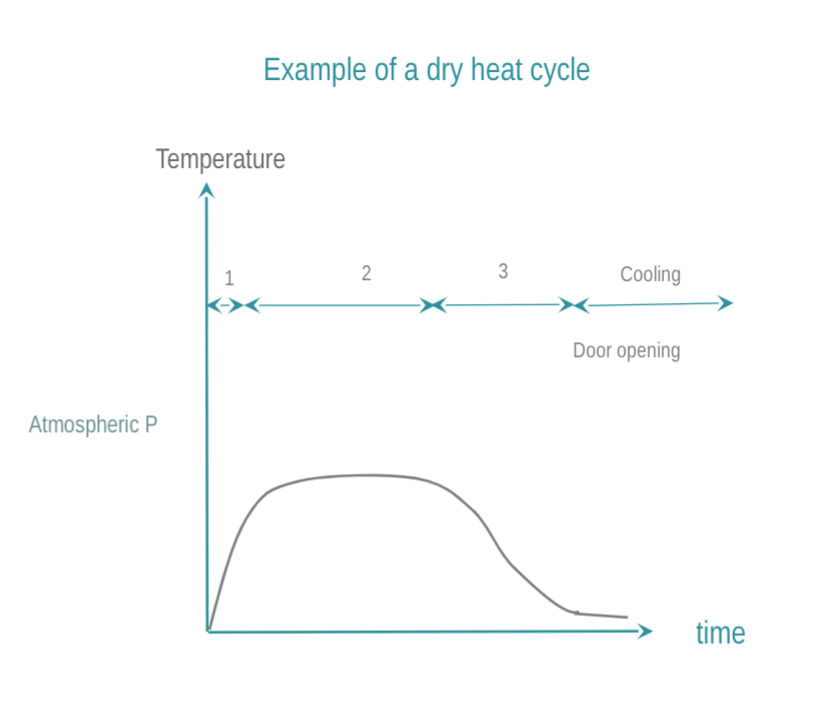
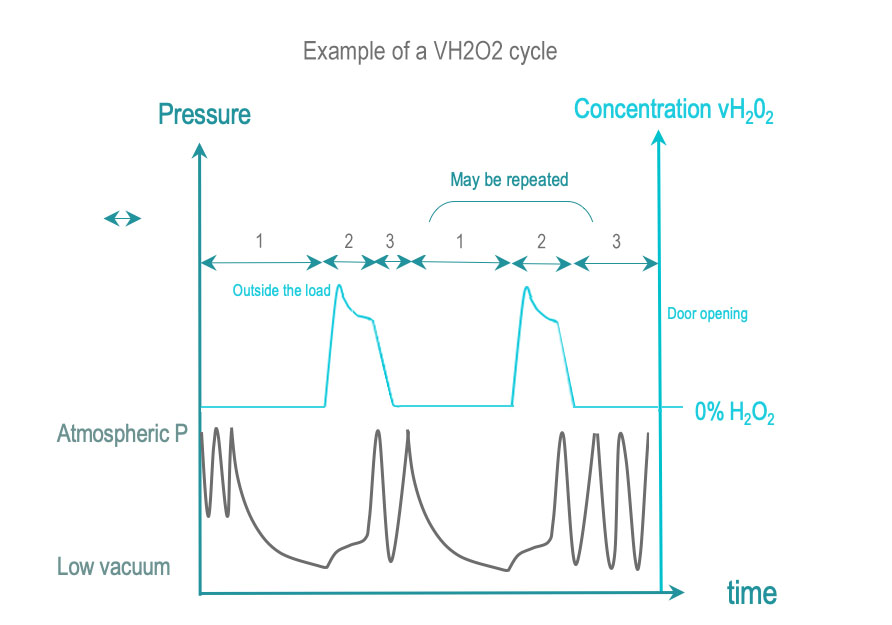
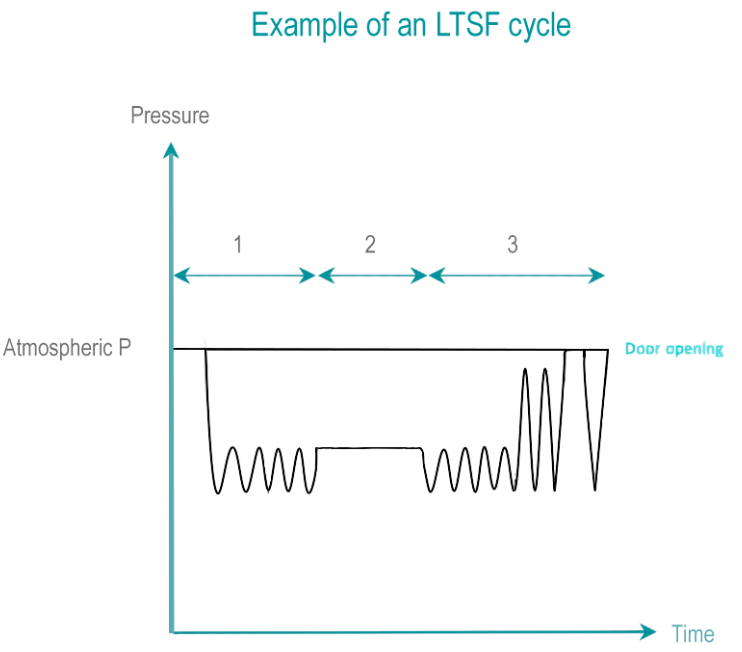
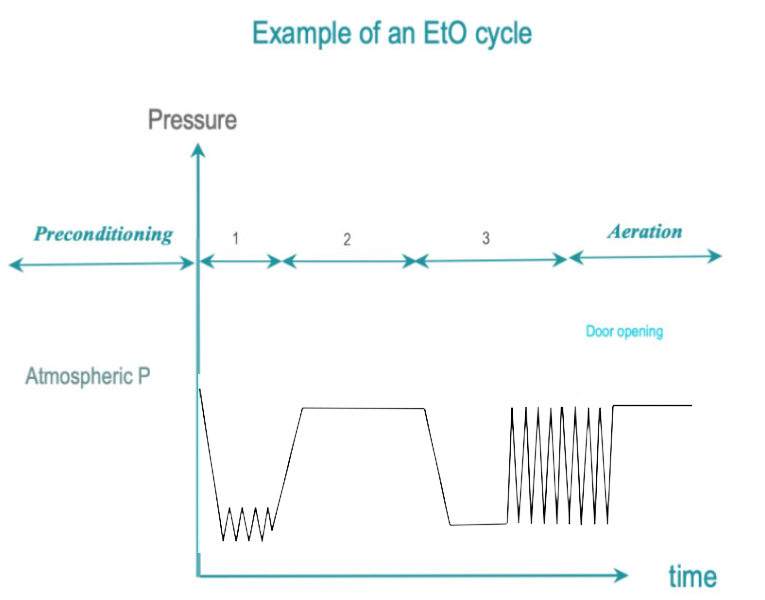
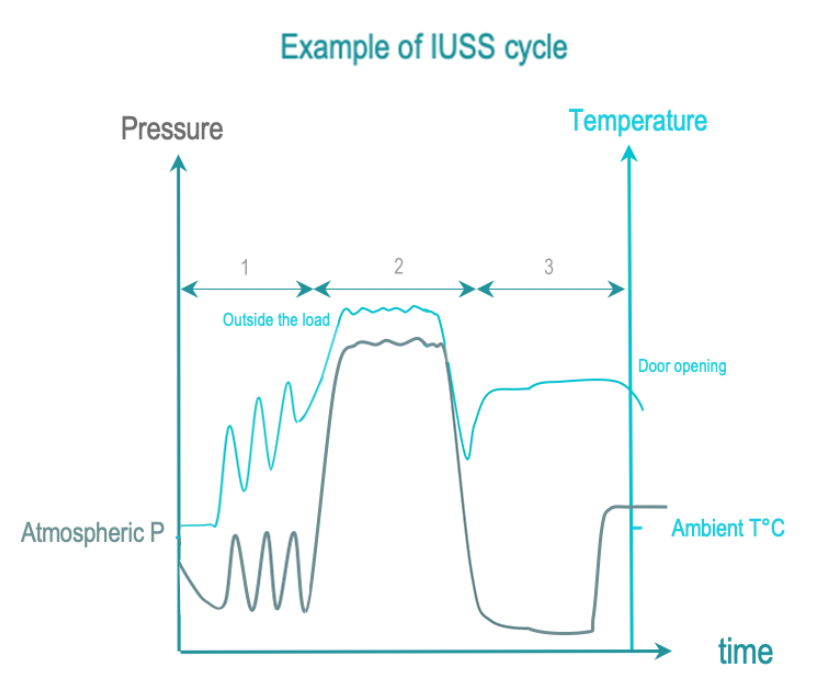
The 3 phases of IUSS are as follows :
- Conditionning: Vacuum and steam injection withdraw air from the chamber and load, and replace it with saturated steam. The load temperature is progressively increased.
- Exposure: The saturated steam injection is completed and given time to diffuse throughout the load.
- Removal : Condensation is withdrawn by vacuum and heating. The load is allowed to cool down after opening the door for safe handling by the operator.
Some countries do not allow IUSS, others may tolerate it. Recommendations usually advise to make an evaluation regarding the need for point-of-use reprocessing. RMD inventory may have to be improved to afford reprocessing by a central sterilization department.
![]() There are no international standards for IUSS.
There are no international standards for IUSS.
![]() Local regulations or guidelines may require or recommend that sterilization takes place in centralized sterilization department thereby prohibiting point of use sterilization. Operating theatres are usually not equipped and organized to clean and dry RMD’s as consistently as a centralized sterilization department.
Local regulations or guidelines may require or recommend that sterilization takes place in centralized sterilization department thereby prohibiting point of use sterilization. Operating theatres are usually not equipped and organized to clean and dry RMD’s as consistently as a centralized sterilization department.

WFHSS key recommendations for sterilization
IUSS to be replaced by steam sterilization
Go to IUSS sterilization →
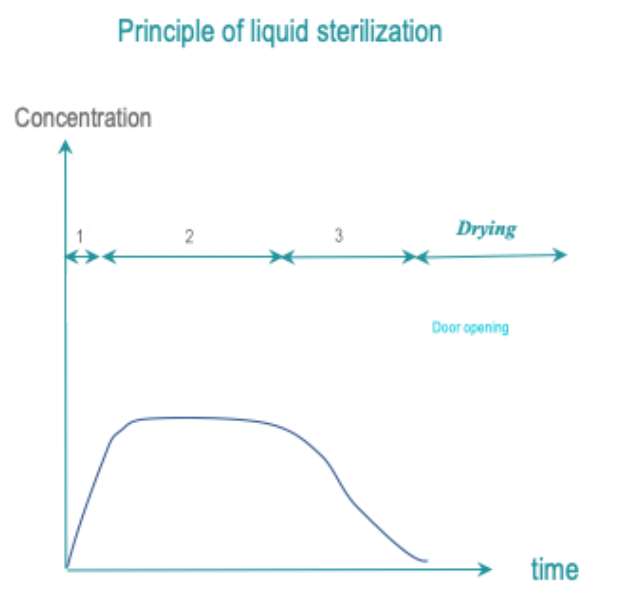
1 of 16 Liquid sterilantTo be evaluated by WFHSS
Go to Liquid sterilization →
2 of 16 SteamSteam 134°C or 132°C preferred when allowed by RMD IFU
Go to Steam sterilization →
3 of 16 LTSFCycle according to RMD IFU
Go to Low temperature steam formaldehyde →
4 of 16 VH2O2Cycle according to RMD IFU
Go to Vaporized H2O2 →
5 of 16 EtOCycle according to RMD IFU
Go to Ethylene Oxide →
6 of 16 Clean, dry, packaged RMDNon packaged for non terminal sterilization
Go to choice of sterilization process →
7 of 16 Sterile Medical DeviceNon packaged RMD for immediate use when non terminal sterilization
Packaged RMD for storage when terminal sterilization
Go to choice of sterilization process →
Terminal sterilization preferred to
non terminal
Go to choice of sterilization process →
9 of 16 Non terminal sterilizationThe RMD is not protected by a packaging and must be immediately used after sterilization
Go to choice of sterilization process →
10 of 16 +Terminal sterilization preferred
Go to recommendation of WFHSS for sterilization →
11 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
12 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
13 of 16 +According to RMD IFU
Go to choice of sterilization process →
14 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
15 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
16 of 16







